LC-MS Based Glycomics Analysis using Orbitrap-based Mass Spectrometers with SimGlycan Software
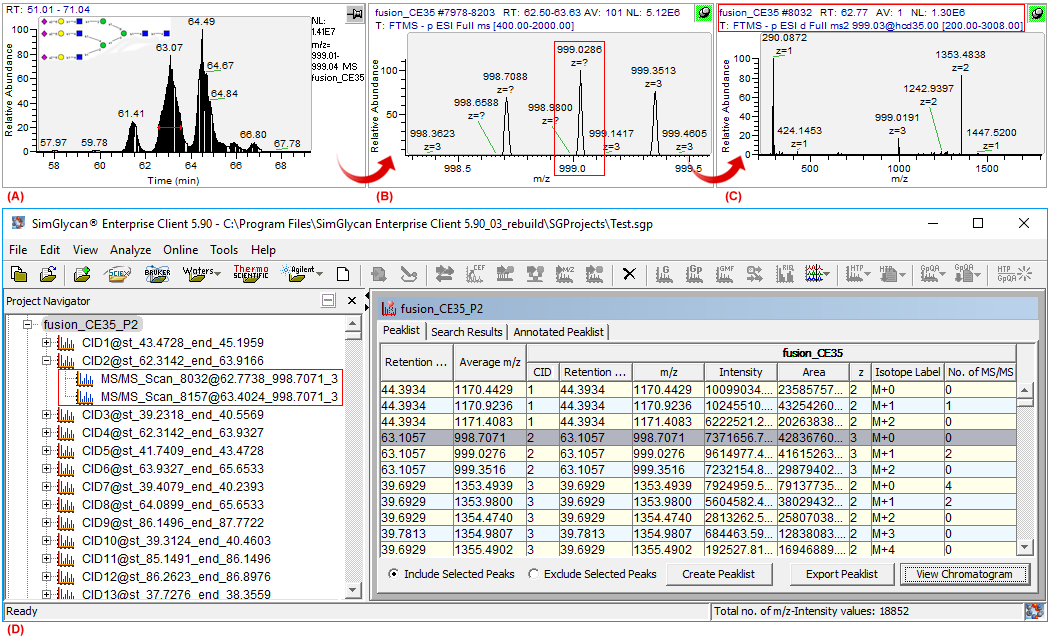 Figure 1: GUIs of Thermo's Xcalibre software showing (A) An extracted ion chromatogram(XIC) of m/z 999.02 along with a glycan structure, one of the probable isomers of (Hex)5 (HexNAc)2 (NeuAc)3, identified by SimGlycan software, (B) Data collated through MS1 scans at FWHM of the XIC: Isotope cluster with base peak at m/z 999.086, the M+1 peak of the cluster, (C) The MS/MS scan with precursor m/z at M+1 peak, and (D) GUI of SimGlycan software showing the corrected precursor m/z 998.7071 for the MS/MS scans facilitating accurate MS/MS database search.
Figure 1: GUIs of Thermo's Xcalibre software showing (A) An extracted ion chromatogram(XIC) of m/z 999.02 along with a glycan structure, one of the probable isomers of (Hex)5 (HexNAc)2 (NeuAc)3, identified by SimGlycan software, (B) Data collated through MS1 scans at FWHM of the XIC: Isotope cluster with base peak at m/z 999.086, the M+1 peak of the cluster, (C) The MS/MS scan with precursor m/z at M+1 peak, and (D) GUI of SimGlycan software showing the corrected precursor m/z 998.7071 for the MS/MS scans facilitating accurate MS/MS database search.
November 28, 2017
The separation of glycans by chromatography prior to MS analysis can reduce sample complexity, minimize ion suppression and increase dynamic range and separation of structural isomers. Rapid development in mixed-mode columns and faster scanning in high resolution mass spectrometers has increased the number of glycans resolved and identified by LC-MS. However, these advancements have not only lead to large data sets, but have also resulted in complex datasets. The datasets include MS/MS data acquired at higher isotopic peaks such as MS/MS data acquired at M+1, M+2, M+3 peaks of the isotope cluster of an LC-compound. Hence, identification of isotope clusters of LC-compounds, identification of their corresponding MS/MS scans, and assignment of correct precursor m/z values from their corresponding isotope clusters become requisite steps to facilitate automated MS/MS database search for glycan identification. The available free software tools for LC-MS data processing provide limited capability to handle such data. Therefore, we have added a new module to SimGlycan, an MSn data analysis software tool, to streamline this process.
The upcoming version of SimGlycan software offers the following key features that not only improves the accuracy of the peak picking, but also speeds up the analysis multi-folds.
SimGlycan vs Others
The raw data was processed using SimGlycan v. 5.90, MZmine v. 2.29, and GlycReSoft v. 2.14 on a laptop (Dell) with the following properties:
Processor: Intel® Core™ i5-5200U CPU @2.20GHz; RAM: 16.0 GB; System Type: 64-bit OS
The table shows a comparative analysis of the performances of the three software tools.
| S. No. | Lipid Class | SimGlycan 5.90 (Beta) | Mzmine 2.29 | GlycReSof 2.14 |
| 1. | File format of raw data | .raw | .raw | MzXML |
| 2. | Data Conversion Software | None | MSFileReader | ProteoWizard* |
| 3. | Peak Detection option | Precursor m/z values of MS/MS scans | All possible LC-peaks (Chromatogram Builder) | Fit MS/MS Features |
| 4. | Time to complete Peak Detection | 2 minutes | 9 hrs 25 minutes | 1 hr 45 minutes |
| 5. | # of LC-Peaks detected | 3,085 | 4,403 | 616 |
| 6. | # of unique LC-compounds after Molecular Feature Finding | 753 | 4403 | 616 |
| 7. | # LC-compounds with unknown charge state - missing isotopic peaks | 0 | 4403 | 478 |
*Any other software tools that can convert Thermo's Xcalibur .raw files into .mzXML format file
The comparison was performed using data acquired from Thermo Scientific™ GlycanPac™ AXR-1 column by a Thermo Scientific™ Dionex™ Ultimate™ 3000 UHPLC instrument with either a fluorescence or MS detector. MS analysis was performed using a Thermo Scientific™ Orbitrap Fusion™ Tribrid™ mass spectrometer in negative ion mode. Detailed information about the LC-MS experiment is available in the poster titled "Development of Bioinformatics Support for High Throughput Isomeric Separation and the Structural Identification of Glycans by LC-MS".
| Comment | Share |
|


